Oncogenes and Cancer Cells
Cancers are a collection of diseases that result from loss of control over cell division (mitosis). Because the cell cycle is controlled by proteins , and proteins are encoded by genes , cancers are genetic diseases.
Despite being genetic in origin, cancer is not usually inherited in as predictable a way as a Mendelian (single-gene) disorder, such as color blindness. Instead, cancer arises as a consequence of accumulated mutations in somatic (body) cells of a individual. This can happen in two ways. In the first method, a person inherits a cancer susceptibility allele (gene variant) from a parent, and therefore has that mutation present in one copy in each cell of the body. If a mutation then occurs in the second copy of the susceptibility gene in one somatic cell, that cell begins to divide uncontrollably. As the cell divides, the offspring cells inherit these mutations and perpetuate the cancerous characteristics. Alternatively, two somatic mutations may occur spontaneously in the same cell. For example, exposure to ultraviolet wavelengths in sunlight, or to cancer-causing chemicals, can cause somatic mutations. The somatic nature of the mutations that underlie cancer is the
Types of Cancer Genes
Mutations of two major types of genes cause cancer. An oncogene increases cell division rate in an inappropriate place in the body or at an inappropriate time in development. The oncogene is a mutated form of a so-called protooncogene, which is a gene that normally causes high cell division rates where it is needed, such as in the fetus or in a wound where cells must be replaced. The abnormal activation of such a gene in time or place causes cancer. Some oncogenes encode transcription factors , whose overexpression then activates other genes that cause the specific characteristics of cancer cells. An oncogene mutation is usually a single deoxyribonucleic acid (DNA) base substitution.
The second type of cancer-causing gene is a tumor suppressor, whose normal function is to halt cell division. Just as a proto-oncogene has a normal function, so too does the unaltered version of a tumor suppressor gene and its encoded protein; it shuts down cell division when it is no longer necessary. Tumor suppressors are critical to development and to the maintenance of organs because they keep cells from dividing uncontrollably. A tumor suppressor mutation is often a deletion. This makes sense, for removal of the gene lifts its normal control of cell division, and a cancer forms.
Many cancers are the culmination of several genetic steps, which may include the actions of both oncogenes and mutant tumor suppressor genes. For example, a type of colon cancer called familial adenomatous polyposis includes deletions of tumor suppressor genes on chromosomes 17 and 18, and activation of an oncogene called ras on chromosome 12.
Activating Oncogenes
Oncogenes arise when proto-oncogenes mutate or are moved to a part of the genome where their expression is greatly heightened. For example, a virus might insert its DNA into a human chromosome next to a proto-oncogene,
Oncogenic cancers can also start when proto-oncogenes are placed near antibody genes, which are highly expressed during infection. In Burkitt's lymphoma, which causes a large tumor to form in the lymph glands near the jaw, infection with Epstein-Barr virus in B cells places a protooncogene on chromosome 8 next to an antibody gene on chromosome 14. The cancer cells of these patients have the telltale chromosomal exchange.
Chronic Myeloid Leukemia and the BCR-ABL Fusion Gene
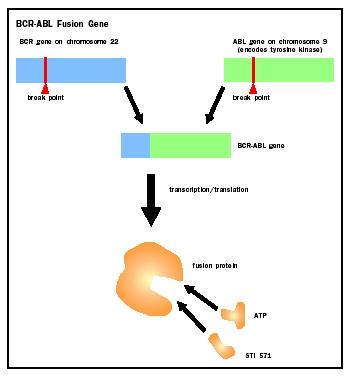
Sometimes a proto-oncogene that has been moved next to a highly expressed gene is transcribed and translated with the second gene. The result is called a fusion protein, which then causes cancer. Discovery of the first cancer-causing fusion protein was a milestone in medical history, and has recently received renewed attention because of development of a highly effective drug treatment. This fusion protein forms in people with a tiny, unusual chromosome, named the Philadelphia chromosome, who also have chronic myeloid leukemia (CML). The errant chromosome forms from a translocation of the tip of chromosome 9 to the minuscule chromosome 22, and transfer of a bit of chromosome 22 material to chromosome 9.
Researchers discovered the Philadelphia chromosome in 1958, when two men were hospitalized in that city complaining of chronic fatigue. Each man's blood had far too many white blood cells, which led to the diagnosis of leukemia. Their blood samples were sent to two young investigators at the University of Pennsylvania, assistant professor Peter Nowell and graduate student David Hungerford, who detected the small, unusual chromosome. At that time, cancer was not considered to be a genetic disorder, and so the apparent association of leukemia with a chromosomal abnormality was a surprise. With time, cases accumulated, and the link strengthened.
By 1972, chromosome banding technology made it possible to describe the nature of the material that makes up the Philadelphia chromosome, and to infer its origin. Janet Rowley at the University of Chicago identified the translocation between chromosomes 9 and 22 that produces the tiny Philadelphia chromosome. By 1984, other researchers discovered the genes that are juxtaposed in the translocation. One gene from chromosome 9 is called the Abelson oncogene (ABL), and the other gene, from chromosome 22, is called the breakpoint cluster region (BCR).
Because the translocation is reciprocal, swapping parts of two chromosomes, two different fusion genes form called BCR-ABL and ABL-BCR. The BCR-ABL fusion gene is part of the Philadelphia chromosome, and this is the one that causes CML. The encoded fusion protein, called the BCR-ABL oncoprotein, is a form of the enzyme tyrosine kinase , which is the normal product of the ABL gene. The cancer-causing form of tyrosine kinase is active for too long, which somehow turns on a cascade of signals that ultimately results in deregulated cell division and cancer. Further evidence that implicates the BCR-ABL oncoprotein in causing CML is that mice genetically modified to harbor the fusion gene develop leukemia. (The other translocated chromosome, which is mostly chromosome 9 material, includes the ABL-BCR fusion gene, which is not known by itself to affect health.)
The discovery that a fusion oncoprotein sets into motion the cellular changes that cause CML has led directly to a new and very promising treatment. The new drug is called signal transduction inhibitor 571, or STI571. (Existing treatment is the immune system biochemical interferon alfa, but it does not help most patients.) In 1990, Brian J. Druker, now at the Oregon Health Sciences University in Portland, proposed to a major pharmaceutical company that he design a small molecule that would lock onto the aberrant tyrosine kinase and squelch its overactivity. He knew that the tyrosine kinase must bind ATP to begin the signal cascade that causes cancer. So he synthesized the small molecule in a shape that enabled it to nestle into the pocket where ATP binds. This, he hoped, would block the signal cascade and stop the cancer.
Following the usual drug discovery trajectory, Druker and his coworkers demonstrated that the molecule indeed halts the cancer in cell culture, and in mice. By 1998, STI571 was ready for phase I clinical trials, which are set up to determine safety at different dosages. In the study, 83 patients for whom interferon had not worked received doses ranging from 25 to 1000 milligrams per day (each participant received the same dose each day). Of the 54 participants who received 300 milligrams or more daily, 53 had their blood counts return to normal, and 29 of them had significantly fewer cells that had the telltale Philadelphia chromosome. In 7 participants, the unusual chromosome disappeared completely.
The new drug appears to have many benefits. Side effects are minimal; the drug is taken by mouth, unlike interferon, which must be injected; response is evident by four weeks; and perhaps most important, it helps people who did not respond to the conventional treatment.
Chronic myeloid leukemia is in many ways a landmark in cancer biology. It was the first type of leukemia to be recognized, in the 1840s, and was the subject of the first chromosome-cancer link. CML was also the type of cancer that led to identification of the first oncogene fusion protein. Finally, a highly effective new treatment is in development, based on understanding the molecular events that underlie the disease.
SEE ALSO Cancer ; Cell Cycle ; Chromosome Aberrations ; Chromosome, Eukaryotic ; Clinical Trials ; Genetic Diseases ; Mitosis ; Mutation
Ricki Lewis
Bibliography
Druker Brian J., et al. "Efficacy and Safety of a Specific Inhibitor of the BCR-ABL Tyrosine Kinase in Chronic Myeloid Leukemia." The New England Journal of Medicine 344, no. 14 (2001): 1031–1037.
Goldman, John M., and Junia V. Melo. "Targeting the BCR-ABL Tyrosine Kinase in Chronic Myeloid Leukemia." The New England Journal of Medicine 344, no. 14 (2001): 1084–1086.
Nowell, P., J. Rowley, and A. Knudson. "Cancer Genetics, Cytogenetics—Defining the Enemy Within." Nature Medicine 4 (October 1998): 1107–1114.
As of early 2001, clinical trials of STI571 had reached more than 2,800 patients in some 30 countries.
Comment about this article, ask questions, or add new information about this topic: